
-
Historical Perspective, Epidemiology, and Methodology
-
Overview of the SCD guidelines and chapters
-
Process and methodology
-
Consensus Statements
-
Clinical Practice Guidelines and the institute of Medicine
-
Prevention of invasive infection
-
Screening for Renal Disease
-
Electrocardiogram Screening
-
Screening for hypertension
-
Screening for Retinopathy
-
Screening for risk of stroke using neuroimaging
-
Screening for Pulmonary disease
-
Reproductive counseling
-
Contraception
-
Clinical Preventive services
-
Immunizations
-
Vaso-Occlusive Crisis
-
Fever
-
Acute Renal Failure
-
Priapism
-
Hepatobiliary Complications
-
Acute Anemia
-
Splenic Sequestration
-
Acute Chest Syndrome
-
Acute Stroke
-
Multisystem Organ Failure
-
Acute Ocular Conditions
-
Chronic pain
-
Avascular Necrosis
-
Leg Ulcers
-
Pulmonary Hypertension
-
Renal Complications
-
Stuttering/Recurrent Priapism
-
Ophthalmologic Complications
-
Summary of the Evidence
-
Hydroxurea Treatment Recommendations
-
Consensus Treatment Protocol and Technical remarks for the implementation of Hydroxyurea Therapy
-
Indications for transfusions
-
Recommendations for Acute and Chronic Transfusion Therapy
-
Appropriate Management/ Monitoring
-
Consensus Protocol for Monitoring Individuals on Chronic Transfusion Therapy
-
Complications of Transfusions
-
Recommendations for the Management and Prevention of Transfusion Complications
-
New Research is Needed
-
Data Systems That Meet the Highest Standards of Scientific Rigor Can Be Invaluable
-
Improved Phenotyping is needed
-
Broad collaborations for Research and Care
-
Beyond Efficacy
-
Look, Listen, Empathize and Ask
Participants 225


Background
Chronic ophthalmological complications of SCD include proliferative sickle retinopathy (PSR) and vitreous hemorrhage. They occur in up to 50 percent of individuals with SCD, and are found more frequently in persons with HbSC disease and HbSS. The presence of PSR is associated with significant visual loss,83 and its peak prevalence in HbSC disease occurs earlier than in HbSS (i.e., about ages 15 to 24 in men and ages 20 to 39 in women).
Ischemia due to vaso-occlusion of retinal arterioles causes the release of vascular tissue factors that stimulate angiogenesis. The neovascular tissue is predisposed to hemorrhage and vitreoretinal traction forces. Although these preretinal neovascular formations are bright red when viable, they appear white following auto-infarction, when they resemble and are called “sea fans.”
PSR is characterized by five stages, beginning with peripheral arterial occlusion and vascular remodeling (Stages 1-11), subsequent neovascularization and sea fan formation (Stage III), and ultimately vitreous hemorrhage (Stage IV) and retinal detachment (Stage V) (exhibit 10). All can be detected by using direct and indirect ophthalmoscopy, slit lamp biomicroscopy, and fluorescein angiography.
Exhibit 10. Stages of Proliferative Sickle Retinopathy (PSR)
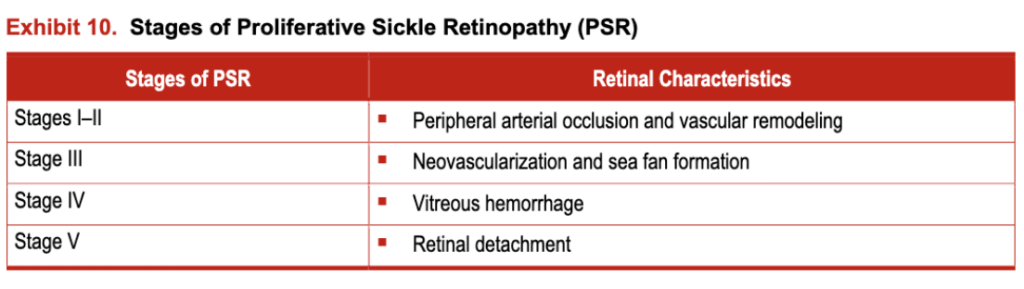
Stages IV and V appear to be more common in individuals with HbSC disease.
Vitreous hemorrhage is a severe complication of PSR360 caused by mechanical stress from trauma or by normal vitreous movement on the delicate neovascular formation growing from the retina into the vitreous chamber.
Spontaneous regression of PSR may occur in about 32 percent of all affected eyes, and lack of progression of sea fans may occur in some people. PSR is commonly managed with laser photocoagulation after consultation with an ophthalmologist. Surgical intervention, including vitrectomy to treat severe vitreous hemorrhage, may be indicated in some people.
Key Question
Summary of the Evidence
Six studies (three RCTs, three observational) and 28 case reports described sickle cell-related acute or chronic ocular complications. The overall quality of evidence for laser photocoagulation was considered high, while the evidence for surgery in people refractory to medical management was considered low.
The three RCTs included 248 people (with likely overlapping populations, the majority of whom were adults) and assessed PSR and the benefit of laser photocoagulation compared to observation. One study reported more than a 50 percent decrease in the rates of loss of visual acuity, and another found that laser photocoagulation was helpful in inducing lesion regression but only in people younger than 25 years of age.
Two of the RCTs reported a significant decrease in the incidence of vitreous hemorrhage, from 45 percent to 4 percent. None of the trials had any form of masking, allocation concealment, or differences in baseline characteristics of the participants.
The three observational studies included more than 140 people, mostly adults, and assessed the roles of laser photocoagulation and surgery in treating sickle cell-related retinopathies. One study found improvement in 83 percent of eyes that received surgery (pars plana vitrectomy) compared to 20 percent spontaneous improvement in the observation arm. One uncontrolled study found lesion regression in 79 percent of treated eyes, with vitreous hemorrhage occurring in only one patient. The last study found benefit from photocoagulation only in “class B” retinopathy (elevated sea fan with hemorrhage). Complications occurred in 13 percent of the untreated people, but not in any treated ones.
Recommendations
Refer persons of all ages withPSR to an ophthalmologist for evaluation and possible laser photocoagulation therapy.
(Strong Recommendation, Moderate-Quality Evidence)
Refer children and adults with vitreoretinal complications of PSR refractory to medical treatment for evaluation and possible vitrectomy.
(Strong Recommendation, Low-Quality Evidence)